Health Happening
The "No-Brainers" for Physical and Mental Health:
CAH is a family of inherited disorders caused by deficiencies in the adrenal enzymes used to synthesize glucocorticoids. There is a lack of CORTISOL hormones, and an increased adrenal production of glucocorticoid precursors and androgens. CAH is marked by acute hirsutism or virilization, sometimes infertility, other signs of masculinity, and possibly stunted height compared to parents.
Mineralocorticoid (primarily ALDOSTERONE) synthesis can also be affected, resulting in electrolyte disturbances, hypotension and syncope (full or partial loss of consciousness).
More than 90% of CAH cases involves an enzyme deficiency of:
- 21α-hydroxylase (CYP21A2 gene deficiency)
And more rarely a deficiency of:
- 3β-HSD (3β-hydroxysteroid dehydrogenase)
or
- 11β-HD (11β-hydroxylase). 8-9 % of CAH cases; occurs one per 100,000 births, but is more common in those of Moroccan- or Iranian-Jewish descent; Resulting mild CAH is much more common, and possibly responsible for 1-2% of female cases of hirsutism and infrequent menstruation. Since 11β-HD deficiency impairs glucocorticoid synthesis, DEOXYCORTISOL accumulates, which may result in hypertension due to the greater-than-physiologic concentrations of DEOXYCORTICOSTERONE causing sodium retention.
ABSTRACT. Over a 39-year period, 38 affected individuals from 25 families were diagnosed. Nineteen families came from Morocco, and in another 2, one parent came from Morocco (80% of all parents). Demographic studies showed that most of their grandparents were born in the region of the Atlas Mountains. In Israel, the overall incidence of the disorder is estimated between 1 in 30,000 to 1 in 40,000 births, but in offspring of Moroccan Jews the ratio is 1 in 5,000 to 1 in 7,000, with an allele frequency of 1 in 70 to 1 in 84 and a carrier frequency of 1 in 35 to 1 in 42.
The clinical expression is characterized by a wide range of variability in the signs of androgen and mineralocorticoid excess. Virilization in the female ranged from enlarged clitoris in the mildest forms, to markedly hypertrophied clitoris with penile urethra and fused labial-scrotal folds in the most severe forms. Hypertension causing vascular accidents and death was observed in both severe and mildly virilized patients, whereas masculinized females were sometimes normotensive.
Based on historical evidence, the origin of the ancestors, and the onomastic analysis of the families surnames, we propose that the mutation of 11β-hydroxylase dificiency in Jews from Morocco may have originated in either the ancient Jewish settlers or the native Berber tribes who lived in the region of the Atlas Mountains in the southern region of Morocco before the destruction of the Second Temple by the Romans, in the year 70 C.E.
New MI. Genetic disorders of adrenal hormone synthesis. Horm Res 1992;37(suppl 3):22-33.
Miller WL. Congenital adrenal hyperplasias. Endocrinol Metab Clin North Am 1991;20:721-49.
New MI. Congenital adrenal hyperplasia. In: DeGroot LJ, ed. Endocrinology. 3d ed. Philadelphia: Saunders, 1995:1813-35.
White PC, Speiser PW. Steroid 11 beta-hydroxylase deficiency and related disorders. Endocrinol Metab Clin North Am 1994;23:325-39.
Migeon CJ, Donohoue PA. Congenital adrenal hyperplasia caused by 21-hydroxylase deficiency. Endocrinol Metab Clin North Am 1991;20:277-96.
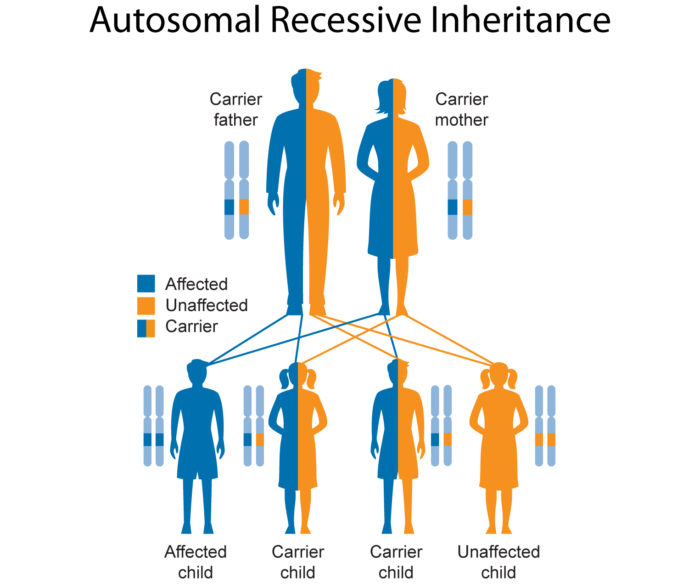
CAH is inherited as any of three main phenotype forms (with clinical presentation given here for females)
(1) Severe form - Classical CAH / "Classic virilizing adrenal hyperplasia" (sometimes called "Salt wasting"CAH). Due to near total deficiencies of 21α -hydroxylase, or 11β-hydroxylase or 3B-HSD, usually detected as adrenal insufficiency in newborn/early childhood, with or without salt-wasting, or later with virilization (masculinization, precocious (early) puberty, hirsutism, acne, amenorrhea); females usually have ambiguous genitalia at birth due to excess adrenal androgen production in utero;
(2) Mild form - "Simple virilizing CAH". Due to deficiencies of 21α-hydroxylase, usually develops iin late childhood; identified by early pubic hair and possibly clitoromegaly, often accompanied by accelerated growth and skeletal maturation due to excess postnatal exposure to adrenal androgens.
(3) Mildest form - Non-classical "Late-onset"CAH (NCAH) / "Non-classic hyperplasia" Due to deficiencies of 21α-hydroxylase and more rarely 11β-hydroxylase, usually develops in adolescence /early adulthood; with signs of androgen excess (E.g. hirsutism, infrequent menstruation, and/or infertility) and possibly shortened stature compared to parents; Girls born with NCAH have normal genitals. This milder form of CAH affects one out of every 100 - 1,000 persons, most commonly due to partial 21α-hydroxylasedeficiency, which is one of the most frequent autosomal recessive diseases.
http://emedicine.medscape.com/article/919218-clinical
A rare form of CAH involves 17-hydroxylase deficiency. Girls appear phenotypically female at birth but do not develop breasts or menstruate in adolescence because of inadequate ESTRADIOL production. They may present with hypertension.
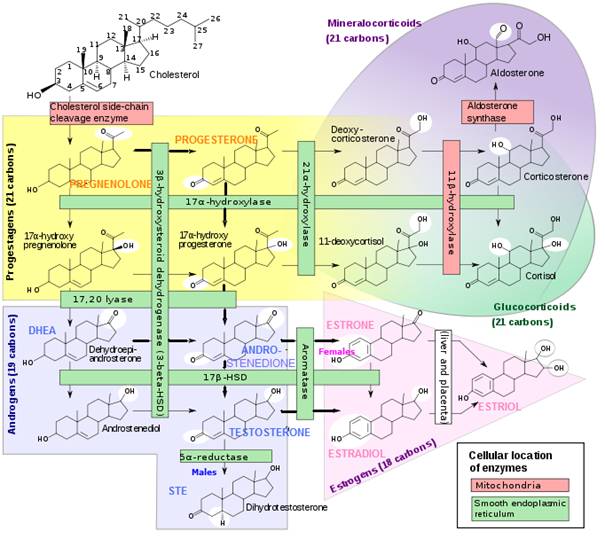
Adrenal androgen synthesis
- The primary adrenal androgens are: dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEA-S) ( < 10% of DHEA and DHEA-S are produced by the testes or ovaries). DHEA is the precursor of other androgens and is theprimary precursor of natural
- Androgens are byproducts of pituitary ACTH (corticotropin)-stimulated CORTISOL synthesis in the adrenal glands. Note that when low CORTISOL levels stimulate ACTH, the higher levels of ACTH indirectly raises levels of DHEA and other androgens and estrogens. However, if the pituitary is NOT producing enough ACTH, then the subsequent low CORTISOL will not be associated with higher androgen and estrogen levels
- Certain ethnic groups are at high risk for NCAH. Ashkenazi Jews, Italians, Yupik of Alaska, Slavs2,3,6 and Hispanics have higher rates of NCAH than the general population.
- Symptoms of NCAH. Vary from person to person and by degree, may come and go throughout life, may worsen over time, often mistaken for premature puberty, may overlap with other disorders making diagnosis difficult, and may include:
Both Females and males
Females - symptoms frequently become apparent shortly after onset of menstruation;
Males
NCAH (mild CAH, Nonclassic CYP21A2 deficiency) should be considered in a patient presenting with. Near-syncope, severe acne, mild hyperpigmentation and a poor response to stress or infections. It should also be considered in female patients with signs of virilism, such as clitorimegaly and hirsutism.
Elevated levels of 17-OHP suggest NCAH related to 21α-hydroxylase deficiency
- A single morning blood test for adrenal steroid levels usually elevated with 21α-hydroxylase deficiency may be sufficient to make the diagnosis. Steroids tested are17-HYDROXYProgesterone, ANDROSTENEDIONE and Testosterone;
A basal, morning serum 17-OHP value > 200 ng/dL (6 nmol/L) (drawn in the early follicular phase) strongly suggests the diagnosis. (> 82 ng/dL in children); A basal, morning serum 17-OHP value > 200 ng/dL (6 nmol/L) (drawn in the early follicular phase) strongly suggests the diagnosis. (> 82 ng/dL in children);
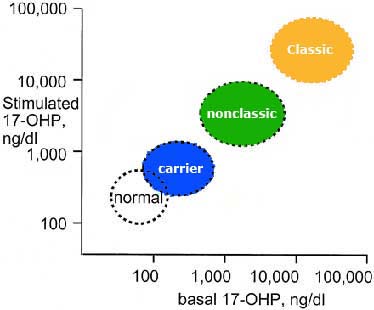
Blood samples are taken before an intravenous dose of ACTH (adrenocorticotropic hormone) and again an hour later. People without CAH respond to ACTH stimulation by releasing CORTISOL into the bloodstream. NCAH blood samples taken after the dose of ACTH show large amounts of 17-OHP, the "raw material"from which CORTISOL is normally made. The results of the ACTH stimulating test are plotted on a "Nomogram", see below, to determine whether the values indicate a diagnosis of CAH.
ACTH Stimulation Test
▪ To perform ACTH stimulation test -serum is drawn to determine baseline 17-OHP and deoxycortisol levels. Administer 250 µg of ACTH intravenously. Wait one hour. Draw post-stimulation serum to determine 17-OHP and 21-deoxycortisol levels;
▪ The diagnosis is confirmed by an exaggerated serum 17-OHP response to high dose (250mcg) ACTH - for most patients serum 17-OHP values 60 minutes after stimulation are typically ≥1500 ng/dL (≥43 nmol/L), and range between 1000 and 10,000 ng/dl (30 and 300 nmol/L)
Elevated levels of DEOXYCORTICOSTERONE (DOC) and 11-DEOXYCORTISOL suggest NCAH related to 11β-hydroxylase deficiency
- Diagnosis tests for DEOXYCORTICOSTERONE (DOC) and11-DEOXYCORTISOL will show elevated levels of these hormones -since 11β-hydroxylase converts DOC → CORTICOSTERONE (precursor to ALDOSTERONE) and 11-DEOXYCORTISOL →CORTISOL.
- 11β-hydroxylase deficiency is characterized by:
Steroid Hormone Conversion Chart
Other abnormalities that may be present with NCAH include:
- High serum concentrations of: 17-hydroxypregnenolone, dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), 3-alpha-androstanediol glucuronide and Progesterone;
- Increased urinary excretion of metabolites of CORTISOL precursors. Particularly pregnanetriol, pregnanetriol glucuronide, (major metabolites of 17-OHP) and 17-ketosteroids (metabolites of androgens, especially DHEA and DHEA-S);
- Elevated DEOXYCORTICOSTERONE/11-DEOXYCORTISOL levels suggest 11β-hydroxylase deficiency
Those with NCAH have insufficient 21α-hydroxylase or more rarely 11β-hydroxylase needed by the adrenal glands to convert 17-HYDROXYProgesterone(17-OHP) into CORTISOL ( and/or ALDOSTERONE production in more severe cases)
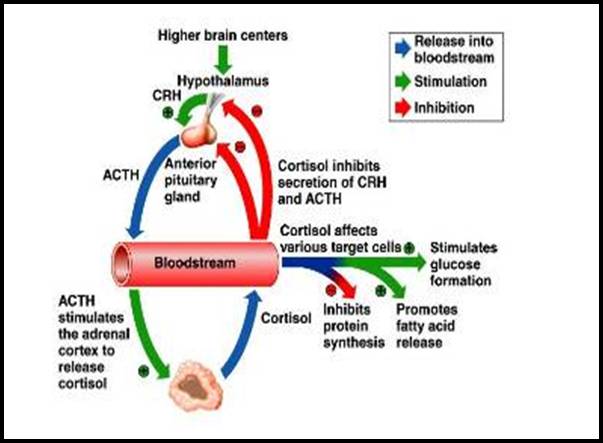
- The adrenal glands are controlled by the pituitary gland (a small pea-sized gland at the base of the brain). Stimulated by the pituitary hormone ACTH (adrenocorticotropic hormone), the adrenal cortex releases hormones, such as CORTISOL and ALDOSTERONE. CORTISOL secretion is ultimately controlled by the central nervous system (CNS) in response to stress, which induces the hypothalamus to release CRH, which stimulates the anterior pituitary to release ACTH.
CORTISOL Release
- CORTISOL ("The Pump-Up"Hormone) is a steroid hormone produced by the adrenal cortex to deal with physical and emotional stress. By putting the body on "Red Alert" and diverting all available energy and raw materials to immediate survival tasks, CORTISOL:
- When the pituitary gland senses insufficient CORTISOL in the bloodstream. It releases ACTH to stimulate the adrenals to produce more CORTISOL, which production is impaired by deficient enzyme activity related to CAH
Some GENERAL symptoms of hypoadrenia/adrenal exhaustion include:
(If you experience 5 or more of these symptoms, you should suspect hypoadrenia and investigate further)
"At-home" Postural Hypotension Test for Hypoadrenia (a.k.a. Raglans test)(Requires blood pressure cuff) Prostrate position. You need to lie down long enough to be in a relaxed state. While lying on your back in a relaxed state, take and record your blood pressure. Normally, with the help of your sympathetic nervous system of which the adrenal glands are a part, your blood pressure will rise 4 - 10 points (mm/Hg) when going from the lying to the standing position. If your blood pressure drops, it may be an indication of hypoadrenia. If your blood pressure drops noticably, you may also feel a little faint upon standing. |
In patients who have few or no symptoms of mild congenital hyperplasia, the risks of treatment may outweigh the benefits
Consultation with an endocrinologist is recommended for patients who require complex hormone regimens.
The following treatment suggestions were found at several internet sites (You should do your own research on this):
Treatment for all forms of CAH requires CAREFULLY MONITORED, LIFETIME glucocorticoid replacement therapy. You will need:
- Glucocorticoids - E.g. hydrocortisone, prednisone or dexamethasone;
Merke DP, Cutler GB Jr. New approaches to the treatment of congenital adrenal hyperplasia. JAMA 1997;277:1073-6.
Laue L, Merke DP, Jones JV, Barnes KM, Hill S, Cutler GB Jr. A preliminary study of flutamide, testolactone, and reduced hydrocortisone dose in the treatment of congenital adrenal hyperplasia. J Clin Endocrinol Metab 1996;81:3535-9.
- Therapy should be at the lowest dosage that achieves prevention of adrenal insufficiency and suppression of excess androgens
Treatment may also require anti-hypertensive therapy








